Pioneering Molecular Innovation:
Multimodal 3D Design with Precise Pose Alignment Crafting Novel Architectures, Rationally Optimized & Pocket-Complementary Compounds.
Unlock Dormant Data:
Synthesize Institutional Insights into Tailored AI Copilots, Powering Context-Aware Drug Discovery.
MolVadoTM Multimodal AI 3D Molecular Generation Model
Pocket-based 3D molecule generation
Substructure-Constrained 3D molecule generation
• Pocket-based 3D molecule generation: Generation of novel and conformationally reasonable drug-like molecules based on the protein binding pocket .
• Fragment-based Molecular Generation: Generation of drug-like molecules based on fixed molecular fragments.
• Rationality Evaluation: Evaluation of the 2D structural and 3D conformational rationality of generated molecules, filtering out structurally unreasonable compounds.
• Activity Molecule Replication: Generation of compounds similar in structure to known active molecules based on the protein binding pocket, ensuring drug-likeness of the generated molecules.
• Support for Online Modification & Iterative Generation: Enabling one-stop modification and labeling on generated molecules, with iterative generation based on selected molecules.

CollectorTM Compound structure & information extraction
• Desktop-based tool, ready to use instantly: Can easily extract molecule structures in only 1s.
• High-precision molecule recognition: Support batch extraction of thousands of chemical structures in patents/documents/pictures from JPG/PNG/PDF formats with real-time correction.
• Flexible recognition and easy export: Can identify both OCSR (chemical structures) and IUPAC (standard chemical names), and enables quick one-click copying or download as SDF/XLS/SMILES/PNG.
![]() Download for Windows
Download for Windows
![]() Download for Mac
Download for Mac
![]() Enter web page
Enter web page
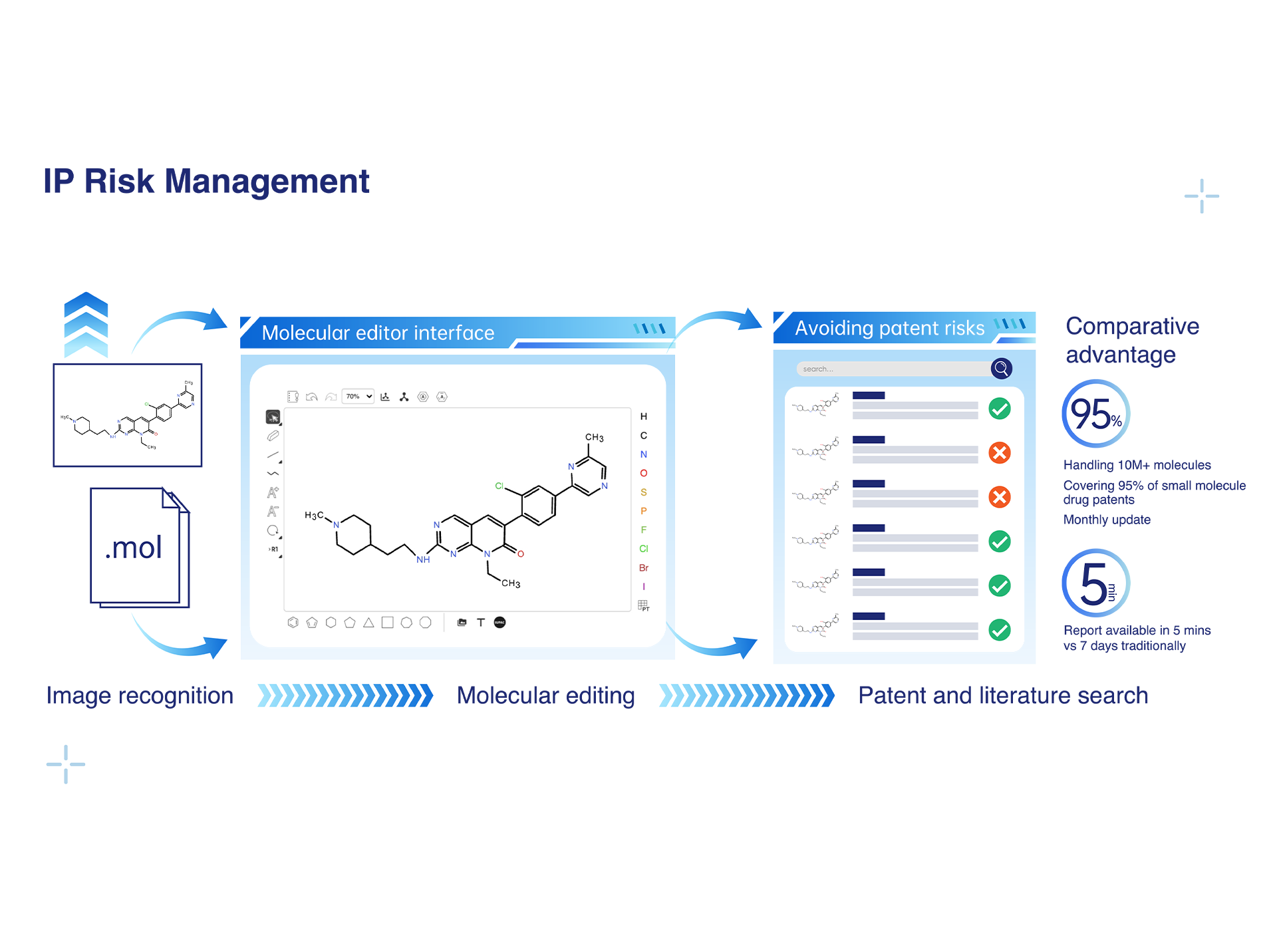
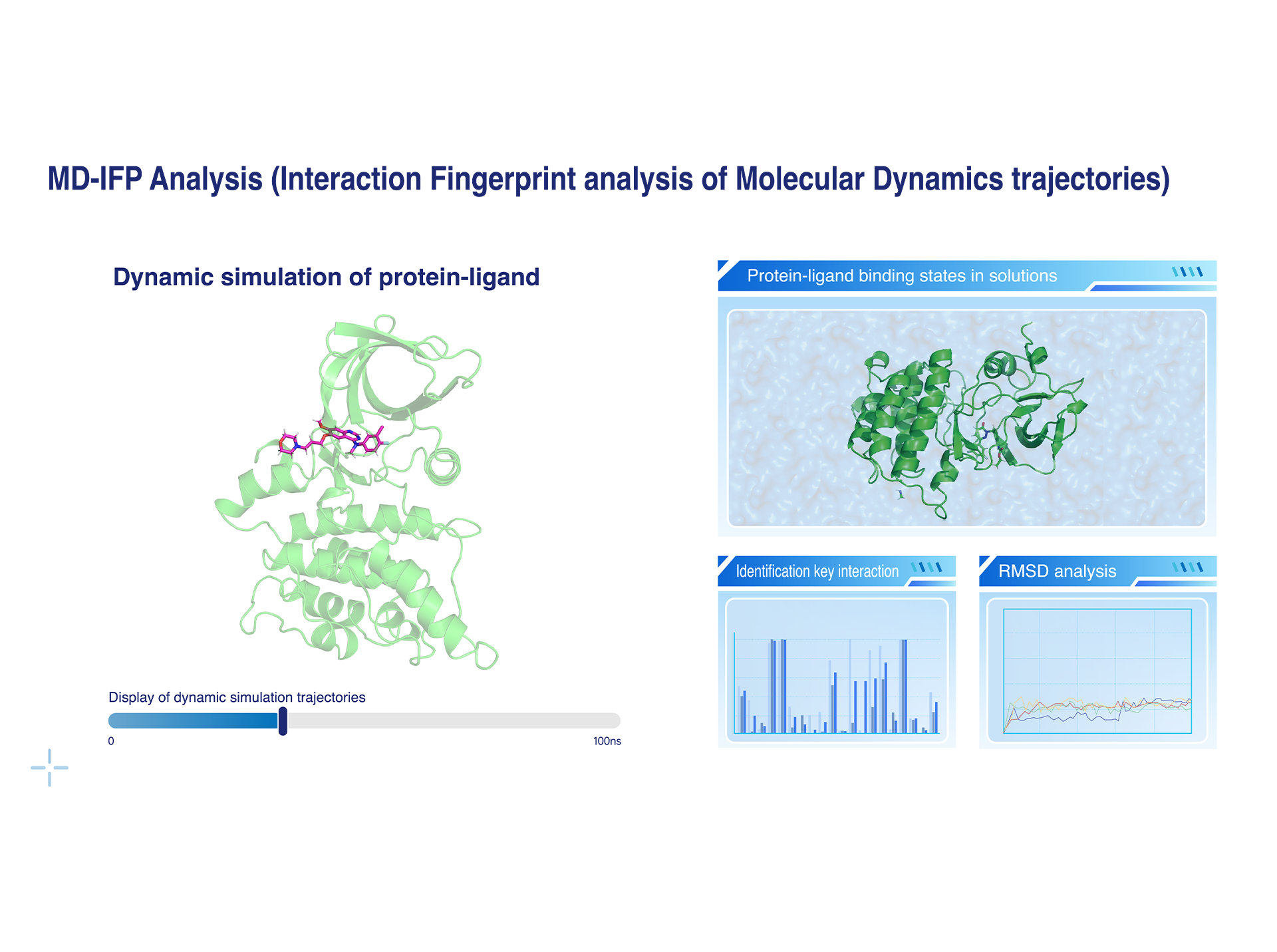
MD-IFP Analysis (Interaction Fingerprint analysis of Molecular Dynamics trajectories)
• Dynamic Simulation of Protein-Ligand Binding States: Overcoming the limitations of static structures, molecular dynamics simulations are employed to explore the protein-ligand binding states, revealing key conformational changes.
• Non-Covalent Interaction Identification: Hydrogen bonds, π interactions, salt bridges, halogen bonds, and other non-covalent interactions are analyzed, with a quantitative assessment of the contributions of critical residues to ligand affinity, providing a reliable basis for structural optimization.
• AI-Assisted Efficient Analysis: Machine learning techniques are integrated to extract key features from large-scale MD trajectories, enabling the automatic identification of the optimal binding conformations.
• RMSD Analysis: This analysis is used to assess the stability of the ligand’s own conformation and its binding stability within the protein pocket.
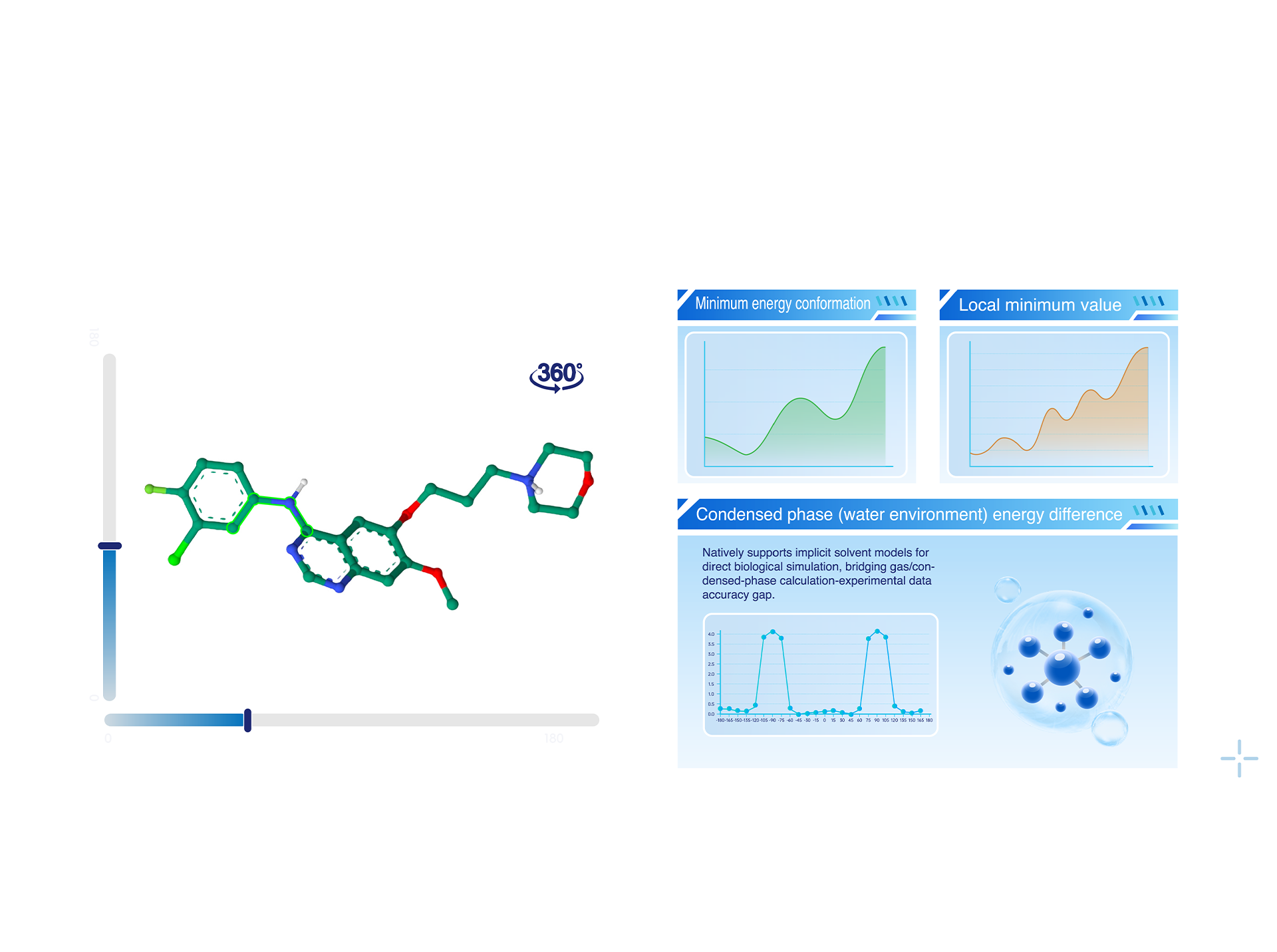
Torsion Scan
• Original Molecular Fragmentation Algorithm: Precisely dissects the compound structure to minimize the impact of structural errors on torsional energy scanning results.
• Quantum Chemistry Precision: Employs double-hybrid density functional theory (DFT) methods, offering torsional energy scans with chemical accuracy.
• Unbiased Conformational Space Exploration: Through a proprietary asynchronous scanning algorithm, it eliminates systematic biases introduced by traditional single-point strategies. Combined with multi-replica molecular dynamics (MD) simulations, it accurately identifies the global energy minimum conformation.
• High-Precision Energy Calculations: Natively supports implicit solvent models, enabling direct simulation of biologically relevant environments, bridging the accuracy gap between gas-phase calculations and condensed-phase experimental observations.
• Torsional Energy Prediction Model: Utilizes machine learning techniques to predict molecular torsional energies, achieving quantum chemistry-level accuracy with the speed of force-field methods.
Substructure Search
• The system extracts and standardizes compound structures from patents and literature to build a database of over ten million small molecule drug structures.
• Patent information coverage exceeds 95%, with more than 95% of small molecule drug patent structures included and continuous monthly updates.
• It supports the generation of results and uploading of structure queries: based on 3D molecular generation results, users can select molecular fragments and perform one-stop retrieval of corresponding patent and literature information.
• The search results comprehensively cover key patent and literature information, including patent publication numbers, application dates, publication dates, current assignees, and literature DOI numbers.